![]()
A Database of Polysachharide 3D structures

| Home | |||||||
| About | |||||||
| User Guide | |||||||
|
X-ray Diffraction |
|||||||
| References | |||||||
| Wiki | |||||||
| Contact us | |||||||
** External Links **
|
|||||||
|
|||||||
Methods of Polysaccharide Structure Determination |
X-Ray Crystallography
......................................................................................... The growth of single crystals which can diffract X-rays to high resolution leads to the possibility of accurate molecular structure determination using crystallographic methods. For all macromolecular entities, the production of these single crystals remains as the single largest obstacle to structure determination by such methods.
More than 50 well-defined polysaccharide X-ray structures, determined by these methods, are reported in the literature. 3-D molecular and fiber structures provide information on shape and the interactions between different polysaccharide chains, as well as with cations and ordered water molecules. This information is vital for understanding the structure-function relationships of polysaccharides. Species studied by these methods include structural polysaccharides: cellulose, mannan, chitin and xylan; storage polysaccharides: amylose, hyaluronan, chondroitin, keratan; gel formers: agarose, alginate, carrageenans, curdlan, gellan and pectate; and finally branched polymers such as galactomannan, welan and xanthan.
Axial and lateral organisation of long-chain molecules is induced (as much as possible) by a process of slow evaporation of a saturated polymer solution. The solution is located between two glass rods which are attached to a fiber puller, the separation distance is increased as a semi-solid state is reached. Alternatively, films are cast by drying concentrated solutions of polysaccharides on Teflon blocks. Orientation is promoted by stretching the films under constant load.
Diffraction patterns are collected by exposure to X-rays under controlled conditions (e.g.. salt saturated helium gas to maintain the fiber and reduce fogging of the photographic film due to scattering by air). The building block of the fiber is the unit cell.
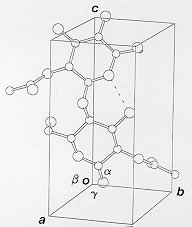
Fig.1 A hypothetical polysaccharide chain whose helix axis is along the The unit cell is of dimensions a, b, c (in Angstrom, Å) and interaxial angles alpha, beta and gamma (in degrees, °). The helix axis coincides with the c-axis, an example is shown in Fig. 1. The nature of the diffraction patterns obtained depends on the degree of order within the specimen, perfect 3-D periodicity gives rise to patterns of spots as shown below in Fig. 2A.
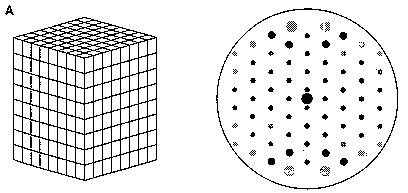
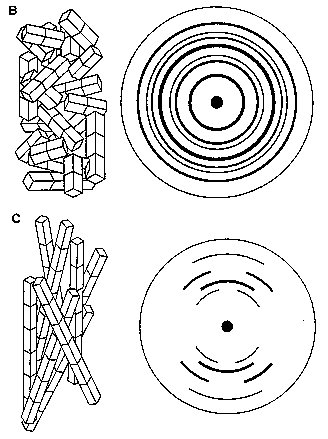
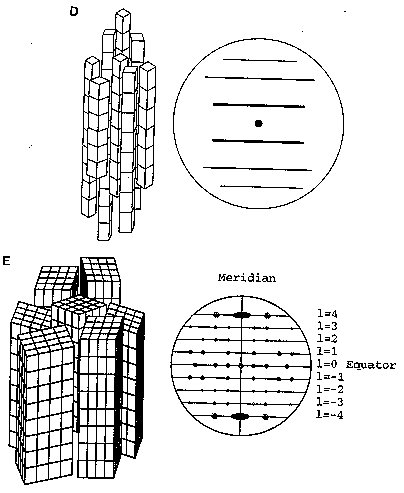
Fig. 2A Different diffracting specimens (Reproduced from Chandrasekaran, 1997) (A) Ordered unit cells produce a series of Bragg diffraction spots. The diminished order found in fiberous samples can be grouped into four major categories (figure 2B-E) (B) An assembly of randomly oriented microcrystallites diffracts to produce a series of concentric rings. (C) An assembly of partially oriented blocks of microcrystallites diffracts to produce large arcs. (D) An aggregate of microcrysatllites whose long axes are parallel, but randomly oriented, diffract to produce a series of layer lines. (E) A polycrystalline and preferentially oriented specimen diffracts to give Bragg reflections on layer lines. The meridonial reflection on the fourth layer line indicates 4-fold helix symmetry.
Fiber-diffraction patterns contain far fewer reflections than single crystals, typically less than 50 reflections going up to at most 3Å. Thus the X-ray data alone is not sufficient to solve a fiber structure. The data-to-parameter ratio can be increased significantly by using existing stereochemical information. The use of standard sugar ring geometry means that only three conformational angles (φ, ψ and χ) are needed to describe the geometry of the helix. Refinement of the structure involves the minimisation of a function containing information on observed and calculated structure amplitudes (for both Bragg and continuous diffraction) along with terms which consider how closely the structure matches optimal geometry. Several possibilities exist for the best molecular model. It may include right and left handed helices, single or multiple helices, with parallel or anti-parallel strands. Each possibility is examined and compared on the basis refinement parameters. With good data, ordered water molecules may be located as well as structurally important cations. The final atomic coordinates are typically to an accuracy of within a few tenths of an Å, with crystallographic R-values of around 0.2.
|
* For more detailed information on electron diffraction please refer to
"Molecular architecture of polysaccharide helices in oriented fibers."
Chandrasekaran, R., Adv. in Carb. Chem. & Biochem., 52, 312-439, 1997.