![]()
A Database of Polysachharide 3D structures

| Home | |||||||
| About | |||||||
| User Guide | |||||||
|
Molecular Modelling |
|||||||
| References | |||||||
| Wiki | |||||||
| Contact us | |||||||
** External Links **
|
|||||||
|
|||||||
Methods of Polysaccharide Structure Determination |
|||||||||||||||||||||||
Molecular Modelling
......................................................................................... Carbohydrates constitute one of the most abundant types of biomolecules occurring widely in all living matter. They function as structural or protective materials and as an energy storage. In addition, carbohydrates perform a much broader biological role
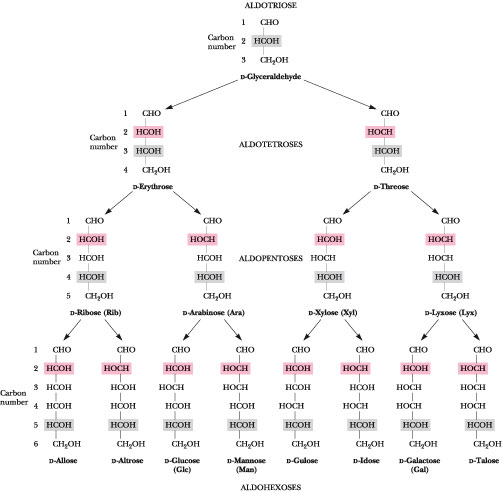
Carbohydrates are polyhydroxy aldehyde, ketone, acid or alcohol compounds that have the general formula (CH2O)n where n is between 4 and 9. Most of the carbon atoms are asymmetric, their various stereo-isomers possess distinct physical and biological properties. Fig. 1 shows the Fisher projection formulae of the naturally occurring D-oses series (the sugars having the absolute configuration R at the last asymmetric carbon atom are called D); the L series of carbohydrate does occur as well, but to a lesser extent. Other natural units are formally derived by simple modification of these sugars such as sulfate and acetate esters, methyl esters of carboxylic acid functions, acetals or ketals of simple carbonyl molecules. For example, in D-glucose the CH2OH group may be replaced by -COOH (e.g. glucuronic acid) CH3 (e.g. fucose) or H (e.g. xylose), an OH group may be replaced by NH2 (e.g. glucosamine) or NHCOCH3 (e.g. N-acetyl glucosamine). Sugar units also occur as simple derivatives. The number of monomeric carbohydrate structures is therefore gigantic.
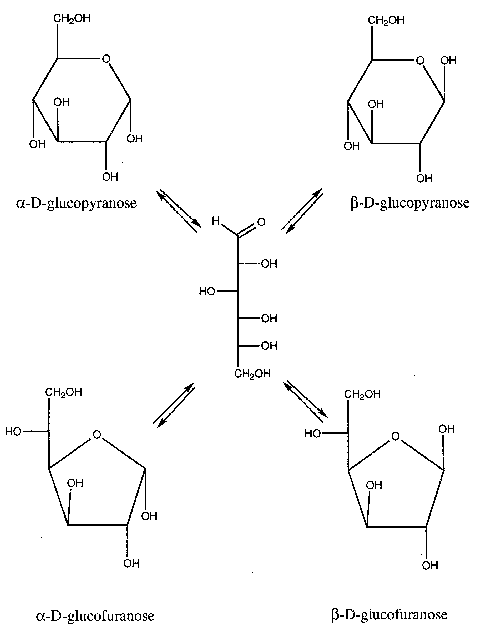
Fig. 1 Fischer Projection Formulae of the naturally occurring D-oses series The open chain forms of monosaccharides are quite flexible, being capable of rotating about each of the single C-C bonds. Because the aldehyde and ketone groups of these molecules are quite reactive, this flexibility often leads to an internal cyclization as the carbonyl group reacts with one of the hydroxyl groups from the other end of the molecule. Depending on the point of attack, the resulting rings can contain five or six atoms, one of which is oxygen; four and seven-membered ring sugars is rather sparse. The closing of the linear molecule to make a ring, creates a new chiral center at C1, called the anomeric carbon. There are two possibilities, that are designated with either an α or β prefix. Several forms of each of the above-mentioned monomers can exist. The cyclization is a reversible reaction, and in aqueous solution an equilibrium between the various forms will exist (Fig. 2). This is called mutarotation.
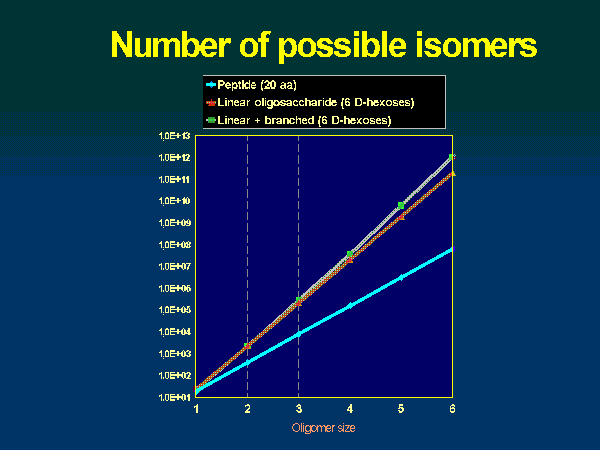
Fig. 2 Mutarotation Monosaccharides can be linked to produce oligomeric structures through glycosidic bond formation. Water is eliminated between the anomeric hydroxyl and any one of the hydroxyls of a second monosaccharide or oligosaccharide. A disaccharide is shown in Fig. 3. The glycosidic linkage consists two bonds, the glycosidic C1-O and the aglycone O-C4 ones. In the case of (1→6) linkages three bonds connect the consecutive sugar rings. Carbohydrates can exist as mono- oligo- and polysaccharides; however, they reach maximum complexity when they are covalently attached to other molecules in glycoproteins, glycolipids, or glycopeptidolipids. The number of possible oligo- or polysaccharides structures is enormous. There are 21 different compounds that can be formed by condensation of two D-glucose units. Since there are multiple points of possible linkage on a monosaccharide residue, branching of the chain is quite a common feature in carbohydrate containing molecules. In theory, the number of all possible linear and branched isomers of a hexasaccharide was found to be more than 1012 (Fig. 4). Not all of these actually occur in nature, but a large number of them does. The naturally occurring polysaccharide structures can be classified into different groups. Simple homo-polysaccharide structures can be linear (cellulose, amylose) or branched (amylopectin, glycogen). Heteropolysaccharides with various degree of branching can be alternating (agarose, carrageenan), block (alginate), complex linear repeat (gellan), complex branched repeat (xanthan), interrupted and branched (pectin). An illustration is given in Fig. 5. The complex oligosaccharides constitute another class of carbohydrate-containing molecules. These molecules generally vary in size from disaccharides to oligosaccharides of 15 to 20 residues, but may be even larger.
Fig. 3 Cellobiose
Fig. 4 The isomeric barrier (Adapted from : Laine, R. Glycobiology 1994, 4, 759-767 )
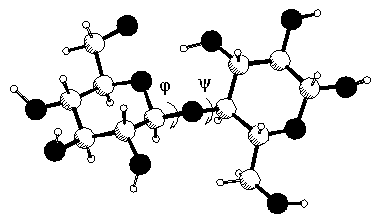
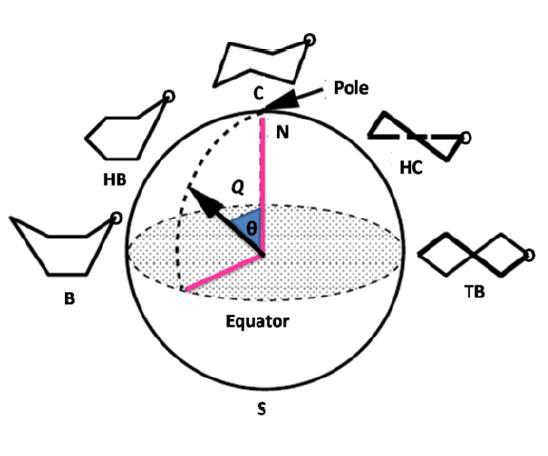
Fig. 5 Examples of different types of polysaccharides. From top to bottom : linear, branched, alternating, block, complex linear repeat, complex branched repeat, and interrupted and branched. Hemiacetal formation between OH-5 and the C1 aldehyde group in aldohexoses (in glucose for example) produces the pyranose ring which contains an endocyclic oxygen atom (O5) and a new asymmetric carbon center at C1. This results in a ring structure having six atoms. The most stable conformations of six-membered ring systems (pyranoses) are the chair forms, but in the majority of naturally occurring pyranoid derivatives one of the two possible chair conformers is considerably more stable than the other. As in the case of cyclohexane ring, the pyranoid ring can also adopt energetically less favorable conformations. Six different skew conformers separated by six different boat conformers, can be identified on the pseudorotational itinerary of a pyranoid ring. The pseudorotation circle of the flexible forms of pyranoid ring and the position of the chair conformers on the conformational sphere of the parameters θ (theta) and φ (phi) are in Fig. 6. Three puckering parameters define unambiguously the position of the individual forms of the pyranoid ring on the conformational sphere. θ is the maximum puckering amplitude, the parameters θ and φ are angles in the range 0° < θ < 180° and 0° < φ < 360°, and can be thought of as polar and azimuthal angles for a sphere of radius θ. The two poles of θ = 0° or 180° represent the energy wheels of the chair conformations 1C4 and 4C1. All twelve flexible forms are located at the equator. In unsubstituted cyclohexane the two chairs forms are the prominent species. Substitution of a heteroatom in the ring and addition of hydroxyls or other exocyclic substituents further stabilize or destabilize ring conformers in relation to cyclohexane. As a general rule, the equatorial position of bulky substituents would be preferred because of a 1,3 syn-diaxial interaction that causes steric clashes. The 4C1 of glucopyranose having all ring substituents in the equatorial position is preferred to the 1C4 conformer in which all the substituents are in axial orientation. However, at high temperature conformational transition to this form can arise spontaneously as demonstrated by the formation of levoglucosan (1,6-anhydro-β-D-glucopyranose). This molecule results from the 1-6 elimination of water from the unusual 1C4 conformation that brings the two hydroxyl groups in close proximity. Besides the trivial case of glucopyranose, the α-L-iduronate ring, a constituent of the glycosaminoglycans heparin, heparan sulfate, and dermatan sulfate, which have potential uses as anticoagulant and antithrombotic agents, exhibits conformational mobility. Three forms, namely 1C4, 2SO and 4C1 of this ring have been suggested to be responsible for the biological activities of these compounds.
Fig. 6 Graphical representation of the Cremer-Pople puckering parameters for a
The five-membered ring forms (furanoses) of D-glucose are not thermodynamically stable. However, very important furanoses molecules are commonly found in Nature. For example D-ribose and D-deoxyribose are found as the building units of nucleic acids, and fructose is a constituent of sucrose. These rings are not planar, either one (envelope form) or two (twist form) atoms are out of the plane that contains the others. The different envelope and twist conformations are of similar energy, and the barrier to their inter-conversions is rather small. Therefore, mixtures of different conformations might be expected in solution. Two puckering parameters are needed to define a conformation for furanoid rings: the puckering amplitude Q and φ the phase angle. Pseudo-rotation energy surfaces of ribofuranose along with its deoxy analogue
The conformations of monosaccharides have been determined by several methods including the formation of complexes from sugars in cuprammonia solution, X-ray crystal structure analysis, infrared spectroscopy, polarimetry and optical rotation. In solution nuclear magnetic resonance spectroscopy is usually the method of choice. The energy of the different ring conformations are affected by the orientations of the hydroxymethyl group. These groups usually exist in staggered positions called gauche-gauche, gauche-trans and trans-gauche. In this terminology, the torsion angle O5-C5-C6-O6 is stated first, followed by the torsion angle C4-C5-C6-O6. It is known from crystallographic studies, NMR measurements, and theoretical calculations that the conformational equilibrium about the C5-C6 bond in aldopyranoses depends significantly upon the configuration at C4. For the 'gluco' configuration (O4 equatorial) the trans-gauche is high in energy and the remaining two conformations are almost equally populated, while the trans-gauche and gauche-trans positions are preferred for those having a 'galacto' configuration (O4 axial). The hydroxyl groups have a high freedom of rotation, and they can participate in the creation of hydrogen bonds. As a result of the many possible orientations of such groups, prediction of the hydrogen bonding network is a difficult task. Most of carbohydrates offer an exceptionally high ratio of hydroxyl groups per saccharide residues. Usually hydrogen bonds are present to either neighboring carbohydrate molecules, glycoproteins, or surrounding water molecules. The extreme diversity of the monomeric units that can be found in carbohydrate structures stressed the need for specific methodologies to facilitate the construction of complex carbohydrates. A carbohydrate fragment library has been created
The anomeric effect describes the axial preference for an electro-negative substituent of the pyranose ring adjacent to the ring oxygen, whereas the exo-anomeric effect describes the rotational preference of the glycosidic C1-O bonds
The exo-anomeric effect influences the rotations around the glycosidic C1-O bond and is therefore important in determining the relative orientations of saccharide units in carbohydrate chains. The exo-anomeric effect is a balance between electronic and steric effects. The three staggered orientations for rotation about the glycosidic bond are not equivalent, the exo-anomeric effect causes preference for the +synclinal orientation of the aglycone group in the α series and -synclinal for the β ones. A review about these effects has been published by Tvaroska
Many force fields for molecular modeling are available. The following force fields are widely used or especially designed for carbohydrates:
The low-energy conformers of a disaccharide can be estimated using molecular mechanics. In such compounds the global shape depends mainly on rotations about the glycosidic linkages, because the flexibility of the pyranose ring is rather limited and the different orientations of the pendent groups have a limited influence on the conformational space of the disaccharide. The relative orientations of saccharide units are therefore expressed in terms of the glycosidic linkage torsional angles φ and ψ which have the definition φ = O5-C1-O-C'x and ψ = C1-O-C'x-C'(x-1) for a (1-> x) linkage. The φ, ψ space can be explored in a systematic way. Both torsions are sequentially rotated in small increments over the full 360° range. At each point of the grid the energy according to the force field in use is calculated. It is then possible to represent the energies of all the conformations available as a contour map in the φ, ψ space. These contour maps enable graphical description of energy changes as a function of the relative orientation of the monosaccharides. They indicate the shape and position of minima, the routes for inter-conversion between conformers, and the heights of the transitional barriers. There are many different methods for calculating contour maps. Calculating potential energy surfaces In the rigid residue, or hard sphere potential surfaces approach, the constituent monosaccharides are assumed to be rigid, with pendent groups fixed. As the φ, ψ values are changed, steric interactions between the pendent groups do occur which are unable to relax. These steric interactions cause a rapid increase in energy. This effect is especially prominent in sterically crowded molecules. In addition, surveys of a large number of known crystal structures along with and supported by semi-empirical calculations reveal small but important variations in pyranoid ring geometries and orientations of pendent groups with the φ, ψ values. These are dependent upon the anomeric and exo-anomeric effects and emphasize the need for a model to include bond length and angle degrees of freedom. The strain produced by steric interactions inherent from rotation of monosaccharide residues is relieved by the inclusion of bond length and angle adjustment in the form of minimization with respect to all degrees of freedom of the system (except φ and ψ) at each grid point. During minimization pendent groups move to the nearest minimum downhill of the starting point. In the process of driving the molecule through unfavorable regions of the φ, ψ space, large steric interactions can sometimes cause pendent groups to overcome torsional barriers. This results in minimization to a different local well. This relaxed map describes a larger accessible potential energy surface than the rigid ones, a lowering of the energy barriers between minima, and a lower energy minimum far removed from the initial starting geometry
Whereas rigid residue maps represent a two dimensional cross section of a 3N-6 dimensional surface, where N is the number of atoms, relaxed maps represent a larger cross-sectional window of a given potential energy surface because it allow minimization of the internal co-ordinates (bond lengths, bond angles and torsional angles) to local low energy wells. However, as minimization will only lead to conformations 'downhill' from the starting structure, the torsional dimension where most conformational variation occurs is limited to only one orientational well. It is possible that rotation of pendent groups over torsional barriers could produce lower energy conformations at that point in the φ, ψ space. Ideally at each point in this space an investigation of all possible combinations of pendent group orientations is required (i.e., assuming that each pendent group can exist in each of the three idealized staggered orientation, 3n different conformations at each point in the φ, ψ space, where n is the number of pendent torsions). This results in 312 (531441) conformations for a simple disaccharide and 319 (1.16 x 109) conformations for a more complex disaccharide typical of heparin. Adiabatic maps attempt to represent the lowest energy of all possible pendent group orientations at each point in the φ, ψ space. On comparison with the corresponding relaxed ones, adiabatic maps are flatter, allow greater freedom about the glycosidic bonds, locate additional minima, and reduce the barriers between the minima. At present there are several different methods for calculating adiabatic conformational maps: In the most commonly method used, the energy at each point in φ, ψ space for several different starting geometries is evaluated systematically and the lowest energy for each point is used to generate the map. This can be very time consuming, so such a systematic search is only possible for carbohydrates of limited size and flexibility. Several procedures have been developed to scan the energy surface as a function of the two glycosidic angles in an efficient way. For example, the Random Molecular Mechanics (RAMM)
The prudent ascent method moves through the φ, ψ space in a way which is dependent on previous minimizations. Large steric interactions are minimized by doing the most favorable geometries first, in a way similar to the local relaxed map. It makes use of inelastic deformations that decrease the energy by recalculating the energies of surrounding geometries using the new lower energy structure as the starting geometry. On average the energy for each point in the φ, ψ space is calculated twice
With the CICADA method (Channels In Conformational space Analyzed by Driver Approach) the potential energy surface is explored by driving separately each selected torsion angle with a concomitant full-geometry optimization at each increment (except for the driven angle)
The Monte Carlo method is essentially a random search method. From a starting configuration (A) a new configuration (B) is generated by random displacement of one or more atom(s). The new configuration is either accepted or rejected, on the basis of an energy criterion. When the energy of B is lower than or equal to that of A, B will be accepted. When it is higher, it will be accepted only if the Boltzmann factor (for the desired temperature) is greater than a random number taken from a uniform distribution between 0 and 1. When B is rejected, A is counted again before a new configuration will be generated; when B is accepted, it will serve as a new starting configuration. The process is repeated many times, and results in a large number of configurations, which should be representative for the system. The method is more efficient for atomic or simple molecular systems than for complex (macro)molecular systems, since a random displacement in the latter case will generally lead to such distortions of a molecule that the energy of a new configuration will usually be very high. Recently, Metropolis Monte Carlo methods have been applied to the conformational analysis of oligosaccharides with the aim of deriving ensemble average parameters
In molecular dynamics simulations an ensemble of configurations are generated by applying motion laws to the atoms of the molecular system. The two major simulation techniques are molecular dynamics in which Newton's equations of motion are integrated over time, and stochastic dynamics, in which Langevin equation for Brownian motion is integrated over time. Several algorithms have been developed for molecular dynamics simulations. Such simulations follow a system for a limited time. Physically observed properties are computed as the appropriate time averages through the collective behavior of individual molecules. For the results to be meaningful, the simulations must be sufficiently long so that the important motions are statistically well sampled. Experimentally accessible spectroscopic and thermodynamic quantities can be computed, compared, and related to microscopic interactions. Such modeling techniques have been applied to a wide range of oligosaccharides. The structural flexibility of these molecules has been confirmed. Highly branched oligosaccharides have also been investigated by means of molecular dynamics. The results obtained with these have been interpreted as demonstrating a more rigid behavior than that found for the linear ones. It should be noted that molecular dynamics is severely limited by the available computer power. With presently available computers, it is feasible to perform a simulation with several thousand explicit atoms for a total time of up to about a few nano seconds. To adequately explore the conformational space it is necessary to perform many such simulations. In addition, it may be possible that carbohydrate molecules undergo dynamical events on longer time scales. These motions cannot be investigated with standard molecular dynamics techniques. It is important to recognize that most quantum mechanical and molecular mechanical procedures are designed to treat molecules in the isolated stale. Omission of the effect of the environment from the calculation results in a neglect of the fraction of the energy contribution that arises from these interactions. For example, a carbohydrate in an aqueous or crystalline environment will usually form hydrogen bonds only to neighboring molecules, while the simulation of the molecule in vacuo is dominated by conformations with energetically favorable intramolecular hydrogen bonds
Several different approaches have been proposed to treat the solvation effects
At present, the best approach is the inclusion of the environment in the simulation, viz. a molecular dynamics simulation with explicit water molecules or other surrounding molecules. By applying periodic boundary conditions a true, but still very small, system is simulated. Of course, this is very time consuming for an oligosaccharide in water.
Probing potential energy surfaces It is the objective of the present section to illustrate that potential energy surfaces can be put to a demanding test. Indeed, many observable properties of oligosaccharides can be calculated and have been shown to be sensitive to the details of the conformational energy surface. More than 3600 crystal structure determinations of carbohydrates are listed now in the Cambridge Crystallographic Data Base. X-ray analysis gives the best data for the conformation of a carbohydrate. Precise atomic co-ordinates are provided, along with an explicitly defined environment. Although the crystalline state is often dismissed as irrelevant to biological processes, comparisons with crystal structures are among the most precise test of modeling available for carbohydrate molecules, provided that packing forces are taken into account. By molecular dynamics simulations of crystal structures both force fields and methods can be validated
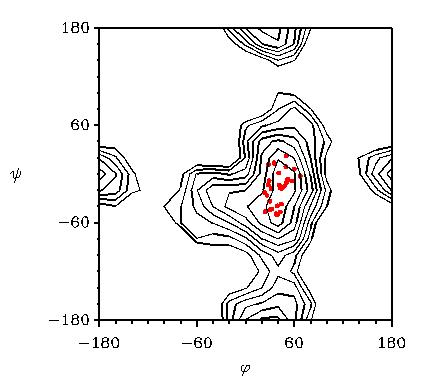
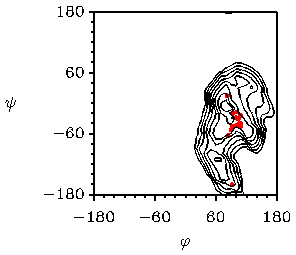
A more common method to use crystallographic data to test computer simulations is the superposition of conformations found in crystal structures on a calculated potential energy map. For example, in Fig. 7 the potential energy surface of cellobiose as a function of the glycosidic torsion angles is given. A search in the crystallographic database for molecules with a link similar to that in sucrose results in a number of conformations, which can be plotted on the calculated surface.
Fig. 7 Potential energy map of cellobiose calculated with MM3
The red dots denote conformations found in crystal structures. Iso-energy contours are 1 kcal/mol apart.
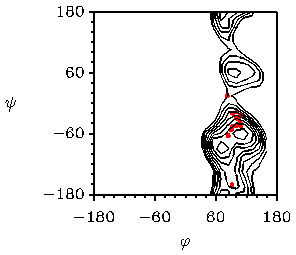
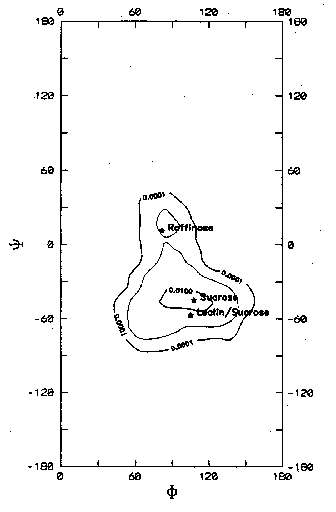
It should be kept in mind that the conformations found in crystals can be influenced by packing effects, so that they differ from the preferred conformation(s) in aqueous solution and in vacuo. A interesting example of the problems that can arise with in vacuo calculations is given by sucrose. In figure 8 maps calculated with MM3
Fig. 8 Potential energy map of sucrose calculated with MM3 (top left )
In solution, the method of choice to study the 3D structure of saccharides is nuclear magnetic resonance (NMR), through the parameters represented by chemical shifts, coupling constants, nuclear Overhauser effects (NOEs) and also relaxation time measurements. While the conformational dependence of the carbon chemical shifts is far from understood, coupling constants can be used to evaluate the magnitude of the torsion angles, nOe measurements can provide estimations of distances between protons located in rather close proximity. In addition, relaxation time measurements give information on the mobility and the behavior of molecules in solution. A major difficulty in the determination of the conformation of an oligosaccharide from NMR data is the flexibility of the carbohydrates, and especially of the glycosidic links. When multiple conformations are present in solution, NMR data will represent a time-averaged conformation. Since the geometrical parameters are usually related in a non-linear way to the experimental data, these data can be very misleading. Consider, for example, an oligosaccharide in solution that occupies two distinct conformations, one with a relatively short distance between the protons at both sides of the link, and one with a rather large distance (which is preponderant). The measured NOE is an average value, so the NOE could easily lead the interpreter to a single non-existing conformation. Even when it is known that two conformations are present, errors can easily be made since the preponderant conformation will produce only a small contribution to the resulting NOE.There are not many experimental means, other than NMR, suitable for probing carbohydrate conformations and evaluating calculated potential energy surfaces. However, the optical activity of saccharides depends on their chemical composition, configuration, and conformation. Models have been developed and applied to disaccharides in aqueous solution
The Disordered State of Polysaccharides The polysaccharide chains in solution tend to adopt a more or less coiled structure. Such a dissolved random coil would fluctuate between local and overall conformations. Polysaccharides are able to assume an enormous variety of spatial arrangements around the glycosidic linkages because these molecules have extensive conformational freedom. Theoretical polysaccharides models are based on studies of the relative abundance of the various conformations, in conjunction with the statistical theory of polymer chain configuration
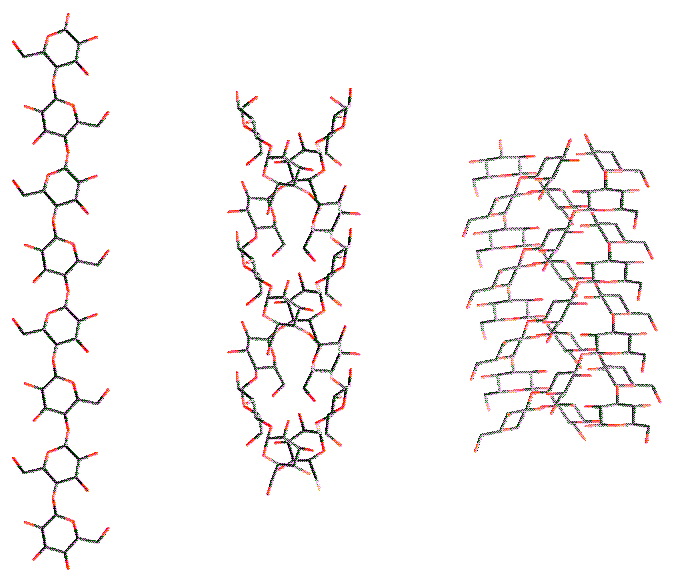
The Ordered State of Polysaccharides Like other polymers, polysaccharides form helical structures. The helix symmetry can be denoted by uv, which means that there are u repeat units in v turns of the helix
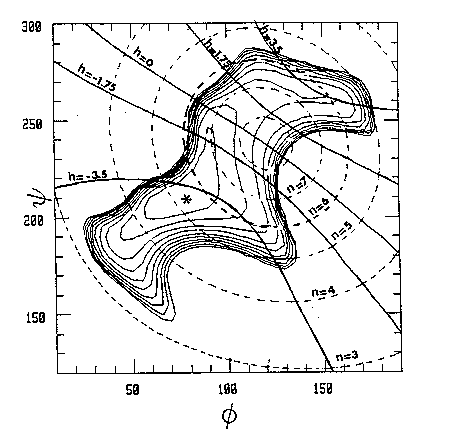
Fig. 9 Examples of a single helix (cellulose), a double helix (amylose) and a triple helix (β-1→3-glucan). The combination of molecular modeling and x-ray data for a linear homopolysaccharide is illustrated by amylose. According to the experimental data, the helices have six-fold symmetry repeating in 2.1 nm. In Fig 10 the potential energy surface calculated as a function of the glycosidic torsion angles of maltose (which is the repeating unit of amylose) is shown. The helical parameters of the amylosic strand generated for each combination of these torsion angles are superimposed on the surface. The conformations that are in agreement with the experimental data are found at the intersections of the contours n = 6 and h = 0.35 nm (for a right-handed chirality) or h = -0.35 nm (for a left-handed chirality). The conformations generating a left-handed helix are near the calculated energy minimum, whereas the alternative conformations appear to be unstable. Therefore, a left-handed model seems to be appropriate
Fig. 10 Iso-n and iso-h (in Å) countours superimposed on the potential energy surface of maltose. The iso-energy contours are drawn by interpolation of 1 kcal/mol with respect of the calculated minimum (*). The h = 0 contour divides the map in a right-handed (h > 0) and a left-handed (h < 0) region. For some polysaccharides diffraction patterns have been measured that are not easily interpreted. Agarose, for example, is a linear polysaccharide consisting of alternating 3-linked β-D-galactose and 4-linked 3,6-anhydro-α-L-galactose units (see Fig 11). Three diffraction patterns have been reported that were interpreted as single helices
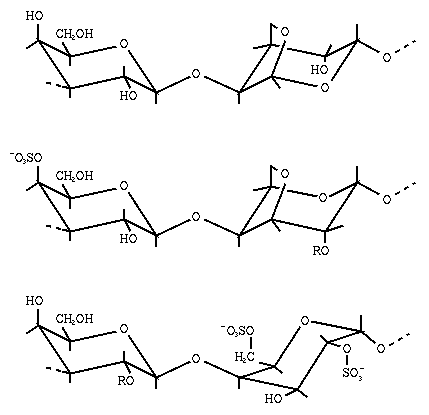
Fig. 11 Schematic representation of the primary structure of agarose (top), iota- and kappa-carrageenan (middle; in the iota-form R = SO3-, and in the kappa-form R = H), and lambda-carrageenan (bottom; R = H or SO3-)
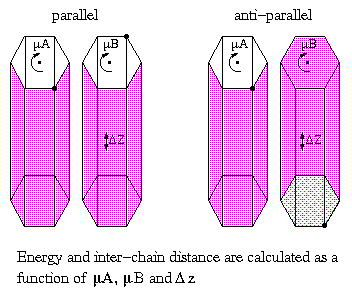
The next step in the determination of the structure of polysaccharides in the ordered state is the investigation of the interaction of different helices, the packing. In a crystal structure of a polymer the chains can be packed parallel or anti-parallel. In cellulose this packing is still one of the remaining questions for the different polymorphs; it is almost impossible to distinguish the two possibilities on the basis of the few reflections that can be measured by X-ray diffraction. Molecular modeling becomes more and more a powerful tool in the study of the packing of polysaccharides. Models can be build and energies can be calculated. A method of calculating chain-chain interactions was published by Pérez et al.
Fig. 12 Schematic representation of the chain pairing procedure.
This procedure has been applied successfully for amylose. Amylose is found as A-type (in cereal starches) or B-type (in tuber starches). In both types the same double helix is found. The chain-pairing procedure was applied to the left-handed double-helical model of this helix, and significant low-energy chain pairings were selected, both parallel and anti-parallel. The most promising was a parallel packing of two helices, where the mutual rotations appeared to be coupled and the translation was half the fiber repeat. The inter-chain distance was calculated to be 1.077 nm. This is in excellent agreement with crystallographic studies 4,5, only the inter-chain distance is slightly larger. In A-type starch it is 1.062 nm and in B-type 1.068 nm. Another, looser, type of interaction is seen in the crystal structure of A-type; this arrangement corresponds to a calculated secondary minimum which is among the low-energy chain pairings
Table 1. Relative energies (in kcal/mol, per repeating unit) of the different packings of the different models.
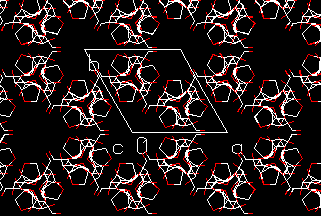
Fig. 13 Predicted crystal structure for one of the observed agarose diffraction patterns (left: top view; right: side view).
With the increasing speed of computers, more complex polysaccharide studies will become possible too. Xanthan, for example, and other charged polysaccharides shall soon be within the possibilities of computer modeling.
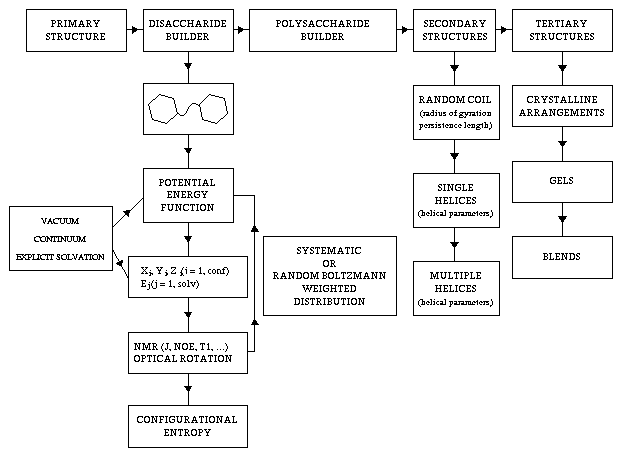
It was the aim of the present work to describe the most recent tools which have been developed for modeling the three-dimensional features of polysaccharides and carbohydrate polymers. It was shown that the primary structures of polysaccharides vary in composition, sequence, molecular weight, anomeric configuration, linkage position and charge density. As a consequence, an almost infinite array of chemical structures and conformations can be generated for polysaccharides. Additional variability also arises from environmental changes such as ionic strength and degree of hydration. In order to cope with such complex macromolecules, the integration of molecular modeling into the biophysics analysis of polysaccharides is required. Steady progresses have been made, which allow a description of the conformations of flexible rings as in the case of five-membered rings, a thorough description of the conformational space which is available for a disaccharide, either in vacuum or in an aqueous solution. Several force fields along with a dedicated parametrization are available. They are in principle capable of dealing with the specific stereoelectronic effects as the anomeric and exo-anomeric effects, or to cope with the enhanced complexity arising from the hydrogen bonding capacities of the carbohydrates. It has been clearly stated that probing in an unequivocal fashion these force fields may not be so straightforward. In particular the use of NMR observable can only be envisaged once the question of internal motions is fully understood. One of the prerequisites for extending the modeling from disaccharides to polysaccharides implies that the rotations of a particular glycosidic linkage can be considered, under some conditions, to be independent of the nearest neighbors. Consequently, the conformations of a polysaccharide can be described conveniently by the glycosidic torsion angles from consecutive dimeric fragments. In solution, polysaccharide chains end to adopt less ordered structures (random coil) that fluctuate amongst different local and overall conformations. Proper modeling can provide insight into the dimensions of these random coils and such descriptors as persistence lengths or characteristic ratio can be readily assessed. Interestingly, the occurrence of local helical regions may be detected from such simulations. It has been observed that these locally ordered conformations are preceding the regular local helical arrangements which are found in the solid state. With the help of such descriptors as helical parameters, the ordered state of polysaccharide strands can be readily characterized. The generation of double or triple helices is then attempted in order to investigate the occurrence of such multistranded arrangements which may be energetically stable. The final step in the determination of the structure of polysaccharides in the ordered state is the investigation of the interactions of different helices which may lead to either the best arrangement(s) between two polymeric chains or to the prediction of the dimensions and the symmetry of a three dimensional lattice The characterization of secondary, tertiary, and quaternary structural levels of organizations of polysaccharides is a prerequisite for understanding the molecular basis of their properties and/or functions and for controlling and manipulating these properties and/or functions through rational changes of molecular fine structure. Therefore, all the different steps which have been described above have been integrated in a general computer program which enables the prediction of the three-dimensional arrangements of polysaccharides chains from the knowledge of the primary structure. Figure 14 is a synoptic of such a program. It incorporates some features such as the prediction of low energy arrangements between different species of polymers chains which could be indicative in the formulation of blends involving polysaccharides. Further work is required, for establishing Quantitative Structure Properties Relationships, in the area of polysaccharides, with a particular emphasis on the gel forming and the viscoelastic properties. The wealth of information available in present data bases can certainly be rationalized. Some of the tools which have been developed should allow automatic searches for meaningful correlations between structures and functions through explorative data analysis. Structure-function or structure property correlations could be then used to model changes arising from structural alterations. This would open the field of polysaccharide engineering.
Fig. 14 Flow chart describing the steps leading from the knowledge of the primary structure to the different structural levels of increasing complexity.
Other challenges will be along the field of modeling supra-molecular structures embedding complex assemblies of polysaccharides. Only realistic modeling of the microfibillar structure of cellulose chains will provide insight in the understanding of the unique rheological properties displayed by such native arrangements plentiful available in renewable raw materials. Other major polysaccharide architecture such as starch granule would also be investigated. Here, the challenge lies in putting together such elementary pieces as double-helices and branching points located at key points of amylose chains to construct the elementary clusters containing over several 100,000 atoms. Only such constructions will lead to the understanding of how the ratio of amylose over amylopectin controls the establishment of unique types of granule. In turns, the control of such ratio, via the routes of molecular biology, will create ad hoc type of granules, for which a significant range of properties can be foreseen. In the area of plant cell walls, the situation is even more intricate since several polysaccharides moieties in the form of a microfibrils and various complex polysaccharides interact via proteins. These are only selected examples which indicate some important structures and architectures as challenging candidates to model within the decade to come.
|
* For more detailed information on electron diffraction please refer to
"Modeling polysaccharides: Present status and challenges."
Pérez, S., Kouwijzer, M., Mazeau, K. & Engelsen, S.B. 1996 in Journal of Molecular Graphics , 14 (6) , 307-321.