![]()
A Database of Polysachharide 3D structures

| Home | |||||||
| About | |||||||
| User Guide | |||||||
| > Pectin Structures | |||||||
| Methods | |||||||
| References | |||||||
| Wiki | |||||||
| Contact us | |||||||
** External Links **
|
|||||||
|
|||||||
Discover Polysaccharides |
Pectin
......................................................................................... Plant cell-wall carbohydrates, which together form the most abundant natural compounds on earth, are our most important renewable natural resource. Omnipresent in agricultural products, these structural carbohydrates play a major role in nutrition. There is considerable commercial impetus behind biotechnological research that is capable of yielding crops with improved mechanical and textural qualities (for example, modification of fruit ripening through transgenic control of polygalacturonase activity). The successful manipulation of cell growth, fruit ripening and texture will depend upon our understanding of how the individual components interact to determine the mechanical properties of the cell wall and of the metabolic pathways which bring about their biosynthesis and metabolic turnover. Structural polysaccharides will also be used increasingly both as a source of energy and as raw materials for industrial processes. In the case of pectins, it is their gelling capacity that has provided the basis for most applications as fine chemicals in the food and pharmaceutical industries (texturizing and thickening agents).
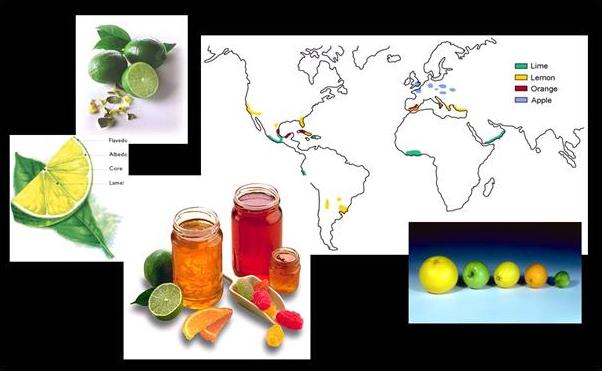
Fig. 1 Pectins : Types, Distribution & Applications
Molecular weight and molecular weight distribution The macromolecular features of pectins vary according to the plant origin, raw material as well as extractions conditions. The heterogeneity of the material along with the complications arising from chains aggregations concur to make the determination of molecular weights and molecular weight distribution challenging exercises. Viscosity measurements are often employed to determine the molecular weight of pectins (Mw), sometime in conjunction with other techniques such as laser light scattering (both at wide and at low angles) and low-speed sedimentation equilibrium analysis. Based on viscometry, the average molecular weight falls within range 50,000-200,000. Number-average molecular weight (Mn) data are obtained from membrane osmometry and end-group analysis. Due to the heterogeneity of pectins, large discrepancies are obtained between Mw and Mn. Other factors being : (i) the polydispersity (ii) the aggregation phenomena (iii) the dependence on the degree and the distribution of the degree of methylation (iv) experimental difficulty during end-group titration. The molecular weight, M may be related to the intrinsic viscosity [η] by the Mark-Houwink equation [η] = 1.4 x 10-6 KMα An α-value of 1.34 for pectin was proposed suggesting a rigid rod-like polymeric backbone. In other works, α-estimates have been reported to be in the range 0.7 - 0.8, for both high-methyl ester (HM-) pectins and low-methyl ester (LM) pectins pointing towards a random coil structure in solution. More recent results obtained, whatever the sources of the pectins, yield a values between 0.8 and 0.9 favoring a slightly stiffer conformation for pectin macromolecules.
The chemical structure of pectins has been the subject of scientific investigations for decades. As the poly-molecularity and poly-dispersity of pectins confer to this polysaccharide strong heterogeneous facets it is not surprising to realize that only recently a 'consensus' description of the primary structure of pectins was proposed.
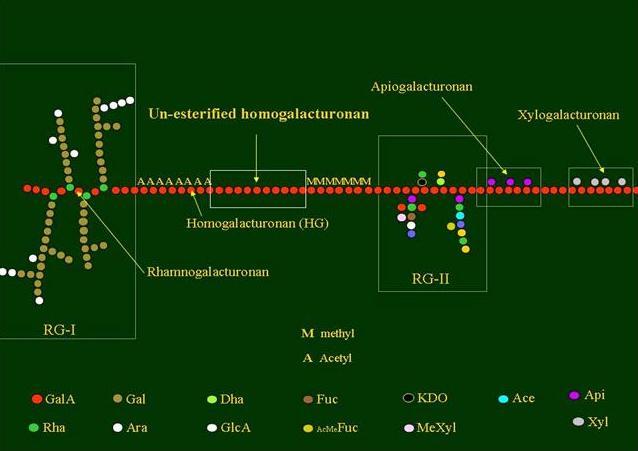
Fig. 2 Building blocks of pectins
The pectin backbone has two main constituents: polygalacturonic acid (PGA) and rhamnogalacturonan (RG I). The PGAs are homopolymers which may contain up to 200 α-D-galacturonic acid residues.
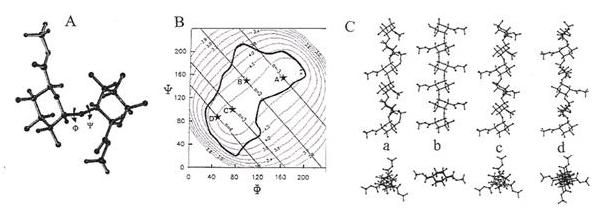
Fig. 3 Studies of a disaccharide model for galacturonan (A) Relaxed potential energy surface of the methyl-esterified galacturonan dimer as a function of the ¼ and ¾ torsion angles. Iso-energy contours are drawn with interpolation of 1 kcal·mol-1 above the minimum of the map. The molecular drawings of the low energy conformers are presented. Iso-n and iso-h contours for the methyl-esterified galacturonic acid dimer are superimposed on the low energy region of the potential energy map (B)Four low energy conformations which would generate integral helices have been indicated by an * and labelled a (n = –3, h = 0.44 nm), b (n = 2, h = 0.43 nm), c (n = 3, h = 0.44 nm) and d (n = 4, h = 0.44 nm). (C)These four helices have been represented using projections parallel and orthogonal to their axis C.
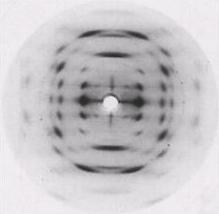
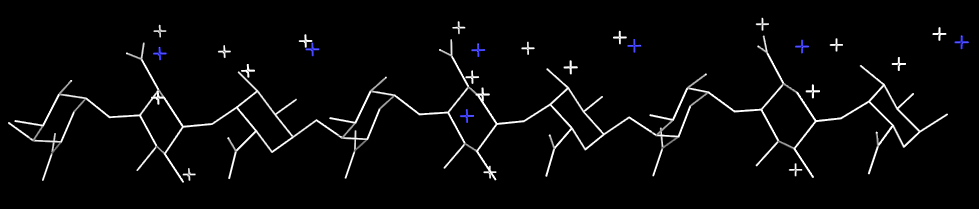
The X-ray diffraction pattern of sodium pectate
Fig. 4 X-ray diffraction pattern of a polycrystalline and well-oriented fiber of sodium pectate showing a 3-fold helix symmetry.
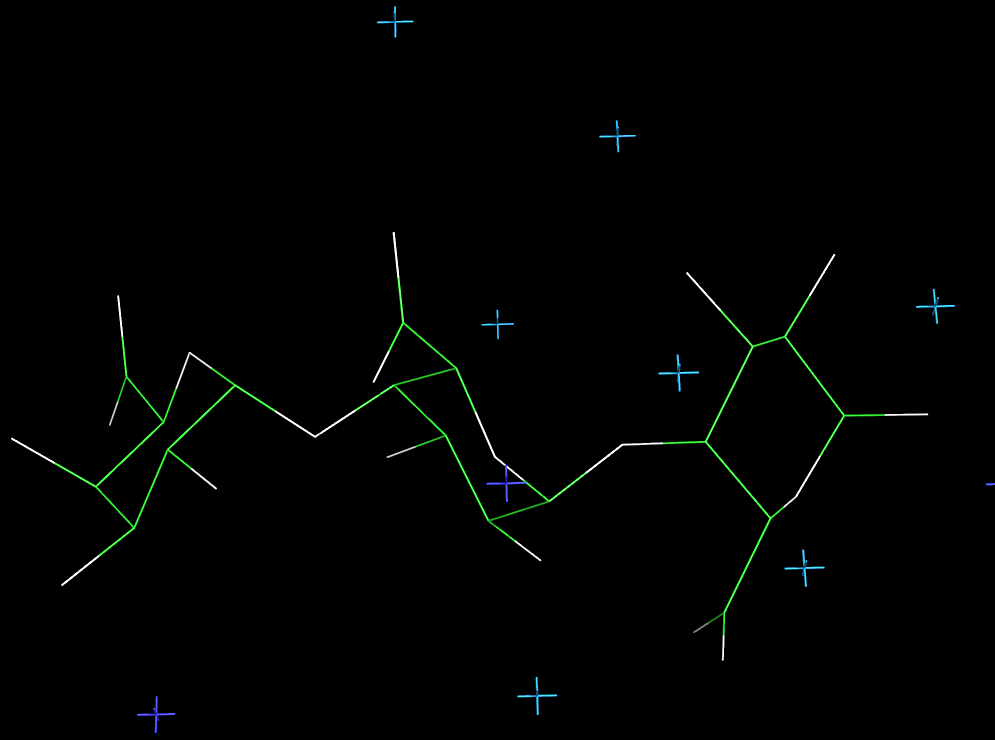
Fig. 5 Representation of the 3D structure of sodium pectate repeating unit
According to the X-ray study
Fig. 6 Representation of the 3D structure of sodium pectate chain
and this interaction is very specific as the polymer exhibits an ability to bind Ca2+ in competition with Na+ over a wide range of relative concentrations. A structural study of all known single crystals of calcium-carbohydrate complexes reported that calcium interactions display considerable stereospecificity and that the strongest binding is likely to result from sets of oxygen atoms that possess the proper spacing and geometry for substituting water in the calcium co-ordination sphere
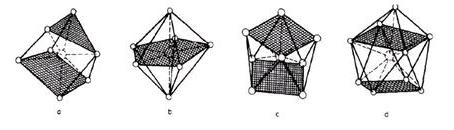
Fig. 7 Computer drawings of (a) a seven-fold coordination of Ca2+, (b) a seven-fold coordination described as a pentagonal bipyramid, (c) an idealized coordinaton geometry found in the eight-fold coordination of Ca2+ and (d) a nine-fold coordination of Ca2+. The two-fold and three-fold helical conformations of polygalacturonate chains were screened for their ability to bind to Ca2+. A Ca2+ ion probe was moved around the van der Waals surface, yielding the location and the amplitude of the most favoured interactions. The two stable helical conformations provide efficient binding to Ca2+. The corrugated polygalacturonate chains offer several oxygen atoms from consecutive residues whose stereochemical arrangement fits into the co-ordination sphere of the cation. The best interaction positions over a 15 kcal.mol-1 window correspond to periodic sites, perfectly defined, with recurrent chelation sites. It is interesting to note that the three-fold helical conformation of polygalacturonate provides an additional binding site. In the two cases, the polygalacturonate chains provide tetradentate chelation sites that are in agreement with those derived from EXAFS experiments for single pectate chains. Nevertheless, as the Ca2+ ion dimerizes the pectate chains in solution, yielding gel formation, the four oxygen atoms predicted as ligands are not at all expected to be involved in the co-ordination sphere of calcium ions at the dimer level. For example, the model derived from fibre diffraction data of concentrated calcium pectate gels mentions that only O-5, O-6, O-2’ from one chain and O-6-O-2’ from the other are co-ordinated to the calcium ions, compared to the four potential O-5, O-6, O-4, O-2’ ligands
Fig. 8 Molecular Model of Pectic Acid A molecular modeling procedure has been developed which involves a pairing procedure that evaluates all the possible associations of the ordered polyuronate chains with calcium ions to form dimers. Starting from the stable ordered forms of polygalacturonate all possible ways to form Ca2+-bridged dimers are evaluated; the parallel and anti-parallel relative arrangements of the chains being also considered. The results indicate that, irrespective of the helical form, the anti-parallel arrangement was the most favorable one. It allowed for an efficient connection of the chains, as illustrated by the shorter interhelical distance, and thus optimized the van der Waals contacts
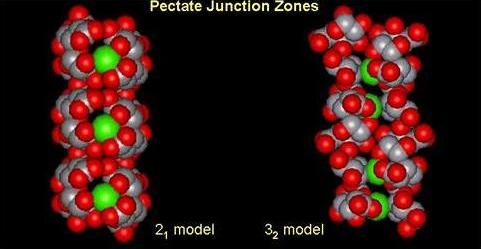
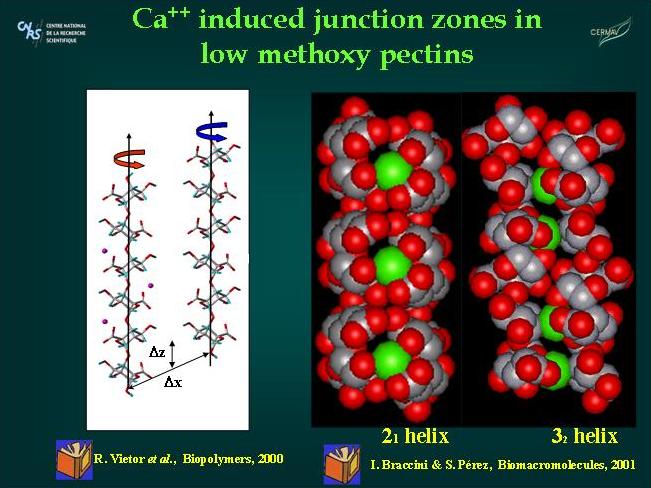
The association of anti-parallel pectate chains with the 2-fold (21) helical conformation provides the best chain-pairing configurations. One chain pairing is particularly favorable as it corresponds to the best configuration for all the individual energy contributions. In particular this structure exhibits an efficient interchain hydrogen bond network with two periodic and reciprocal hydrogen bonds O6…O3, 2.78 Å, and O5…O3, 2.67 Ås that alternate with the calcium interaction sites.
Fig. 9 Molecular Models of Pectic Acid helices and junction zones
The dimers are thought to be strongly associated structures that govern gelation and if sufficient calcium is available, subsequent dimer-dimer aggregation may occur. Several authors have observed that these subsequent associations are easily disrupted by competing ions while the initially formed, 2-chain dimers are not. Therefore, the one issue that remains is why the association between dimers is weaker and more tenuous than that between participating chains within dimers. With respect to this question, the dimer-dimer interaction has been investigated using the best chain pairings obtained for galacturonate (best antiparallel dimer) chains. It was found that the favorable dimer-dimer associations exhibited a different behavior from that of the chain-chain associations. Chain-chain associations are strongly specific while dimer-dimer associations are not. There are roughly two favorable types of chain-chain association yielding dimers. However, dimer-dimer associations occur with many different geometries. In these associations, the energetic contributions exhibit significant variations from one structure to another. In most of the energetically favored systems, electrostatic interactions dominate the energy partitioning with little or no contribution arise from hydrogen bonding. Both the number of carboxylate groups (twice that of an isolated chain) and the geometry of the dimmers allow for numerous arrangements for efficient COO-- Ca2+--OOC electrostatic interactions.
Fig. 10 Dimer formation and interaction with calcium ions
The theoretical results confirm the experimental observations and corroborate the two-stage process that was proposed: for Ca-induced gelation: (1) initial dimerization corresponding to strong associations (2) subsequent aggregation of these dimers. The most convincing argument for the preferential formation of dimers came from competitive inhibition studies where the addition of poly galacturonate chain segments dramatically reduced the observed gel strength. Chain-chain interactions lead to strong dimer associations with important contributions from the van der Waals and hydrogen bonding interactions and for which calcium ions have specific positions in well adapted cavities. The subsequent association of dimmers involves weaker associations that display no particular specificity and seem to be mainly governed by electrostatic interactions.
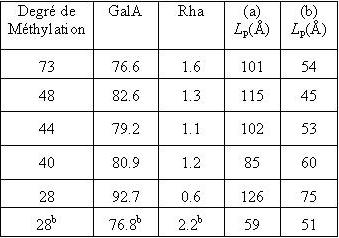
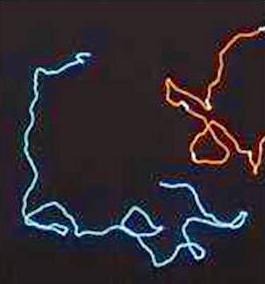
The disordered structure of the homogalacturonan moiety The persistence of these ordered helical structures in solution is not likely to occur; it is therefore of great interest to model the disordered state of polysaccharide backbones and investigate the influence of any given structural motif upon the overall shape of the macromolecule. The characteristic features of the homogalacturonan region of pectins in their random conformation were simulated independently. An extended chain conformation having a persistence length in the vicinity of 10 nm corresponding to about twenty monosaccharide units) was predicted. The introduction of methyl-esterified groups to the galactopyranurosyl residue was also performed following a random distribution along the chain and no significant perturbation was noticed. It can therefore be concluded that the homogalacturonan chains adopt extended conformations having a low probability of folding, a feature that may be largely attributed to the axial-axial nature of the glycosidic linkage. Due to steric hindrance, the conformational (φ,ψ) space is limited and all the conformations are found in a single low energy region. Furthermore, all low energy conformers in this single energy-well generate extended conformations. The homogalacturonan macromolecule therefore tends to adopt a pseudo-helical conformation even in its disordered state. Simulations were also performed to investigate the influence of the insertion of l-rhamnosyl residues on the configurational properties of pectin chains, using the results presented above. Several simulations were performed with increasing amounts of l-rhamnose residues. It was found that, irrespective of the percentage of rhamnose insertions, within the statistical chain remained almost unchanged with respect to those found for homogalacturonan. However, the persistence length is slightly smaller for rhamnose contents in the range 0–15 % and, for example, is decreased by less than 10 % (having a value of 115 Å) when up to 25 % of rhamnose insertion is included in the main chain. This clearly shows that the rhamnose insertion may result in local perturbations but does not induce any change in the global orientation of the main chain. The results derived from molecular modeling confirmed with experimental data on the solution behaviour of pectin polysaccharide as investigated by small-angle neutron scattering and viscosimetry along with a NMR study of the dynamic single stranded conformation of sodium pectate in solution. Therefore, the extended overall chain conformation remains practically unchanged as a consequence of the self-cancellation of the effects of successive paired rhamnose residues in the rhamnogalacturonan I moiety of the pectic backbone. 80 kD > MW > 190 kD
(A) (B)
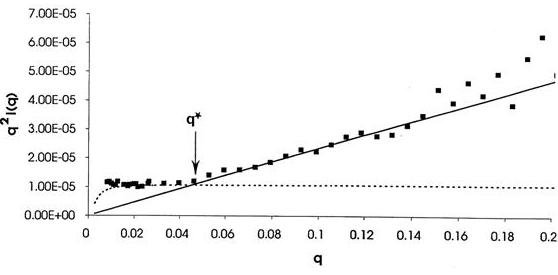
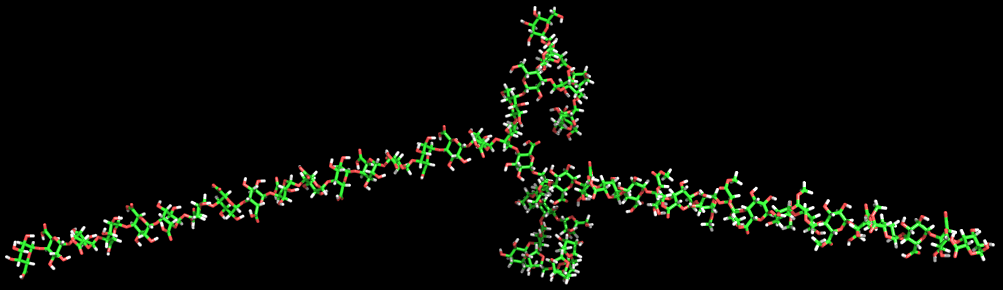
(C) Fig. 11 Solution conformations of pectin polysaccharides (A) Comparison of the determination of persistence length by (a) viscosimetry and (b) neutron scattering of pectins as a function of the degree of methylation and GalA and Rha content. (B) Computer representation of the structure of a pectin chain as generated from Monte Carlo simulations. (C) Determination of the persistence length (Lp) of a pectin sample C48 from small angle neutron scattering data using a Kratky plot. The disordered structure of the rhamnogalacturonan moiety RG-I is a heteropolymer of repeating (1→2)-α-l-rhamnopyranosyl and (1→4)-α-D-galactopyranuronic acid disaccharide units. The length of RG-I segments is not clearly defined but they are usually considered to interrupt long sequences of PGAs. The rhamnose residues also act as sites for further glycosylation in the hairy regions. The percentage of rhamnose residues in pectins is generally very low but can reach up to 35% in some cases.
Fig. 12 The modelled chain of the rhamnogalaturonan moiety.
Fig. 13 The 3D structure of Arabinan
In contrast to the arabinans, the lateral chains of the arabinogalactan type I are orthogonal to the rhamnogalacturonan backbone resulting in a persil mill structure
Fig. 14 The 3D structure of type I Arabinogalactan
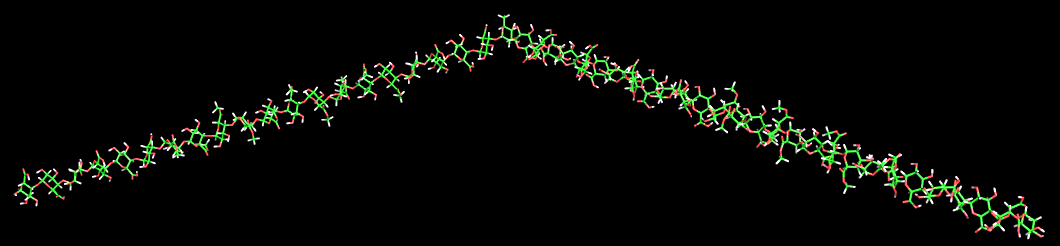
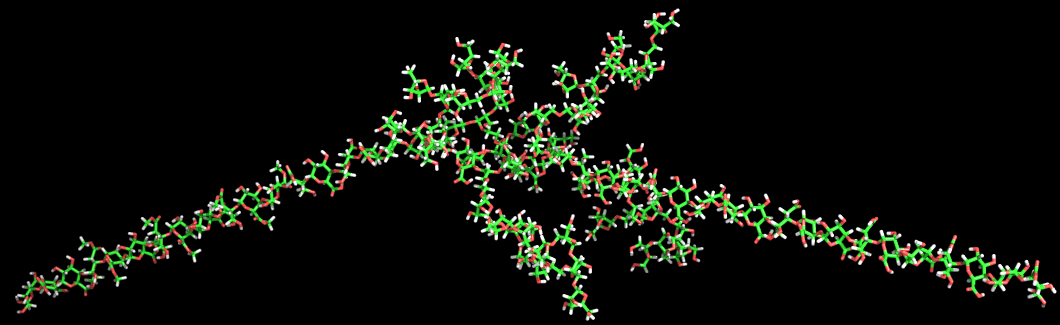
Type II arabinogalactans. The type II arabinogalactans are the most complex of the pectic side chain structures. In contrast to type I arabinogalactans their main branch is composed of a (1→3)- linked -D-galactan chain. This chain is substituted by short (1-6)-linked b-d-galactan chains, which in turn carry additional branches of (1-3)- and/or (1-5)-linked α-l-arabinofuranosy1 residues. The composition of the primary structure selected for modeling corresponds to that proposed by Darvill et al. The tertiary structure of the type II arabinogalactan displays several interesting features. The lateral (1→3)-linked β-D-galactan chain has a fairly open helical structure with a low symmetry (10- fold). The extension per dimeric unit is 9.47 Å. The attached (1→6)-linked β-D-galactan side chains are almost nonhelical linear structures with an extension of 9.5 Å per dimeric unit. The 6-substitution on the (1→3)-linked /3-D-galactan causes the (1β6)-linked β-D-galactan to point away from both the backbone and the (1→3)-linked /3-D-galactan. The combination of the lateral open helical (1→3)-linked /3-D-galactan chain and the almost linear (1→6)-linked -β-Dgalactan side chains makes for efficient packing so much so that even more densely branched structures could be accommodated without steric clashes. The modeling predicts that there will be no steric hindrance even when infinitely long (1-3)- linked β-galactan side chains are substituted by infinitely long (1→6)-linked β-D-galactan side chains, which in turn are substituted by (1→3)-linked α-l-arabinofuranoses.
Fig. 15 The 3D structure of type II Arabinogalactan
|