Agarose
.........................................................................................
Introduction
Agarose is a structural polysaccharide found in certain genera of the Rhodophyceae group of red sea weeds  It is a linear, neutral polymer whose idealized structure is made up of alternating residues of a 1→3 linked β-d‑galactopyranose and 1→4 linked 3,6‑anhydro-α-l‑galactopyranose (Fig. 1). The polymer exists as random coils in dilute or hot solutions, but undergoes a conformational transition to form ordered helices in the solid state . The individual helices are thought to be stabilized by extensive aggregation . Already at low concentrations, agarose is able to form gels. It has been postulated that « structural » impurities found in native agarose, such as the absence of the 3,6‑anhydro bridge or the presence of methyl or sulfate esters, prevent the perfect ordering and introduce kinks in the helices. These kinks might be the cause of gel formation instead of precipitation .
It is a linear, neutral polymer whose idealized structure is made up of alternating residues of a 1→3 linked β-d‑galactopyranose and 1→4 linked 3,6‑anhydro-α-l‑galactopyranose (Fig. 1). The polymer exists as random coils in dilute or hot solutions, but undergoes a conformational transition to form ordered helices in the solid state . The individual helices are thought to be stabilized by extensive aggregation . Already at low concentrations, agarose is able to form gels. It has been postulated that « structural » impurities found in native agarose, such as the absence of the 3,6‑anhydro bridge or the presence of methyl or sulfate esters, prevent the perfect ordering and introduce kinks in the helices. These kinks might be the cause of gel formation instead of precipitation .
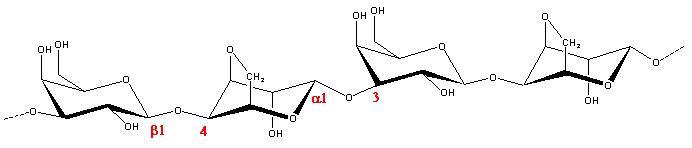
Fig.1 Representation of the agarose repeating unit
Different reports have been published about the nature of the helical structure of agarose. Arnott et al. studied eight different agarose derivatives by optical rotation, X‑ray diffraction and computerized molecular model building methods . They proposed a parallel double-helix model with a 9.5 Å axial periodicity and an internal cavity of 4.5 Å. In this model, the single chains are left-handed three-fold (n = –3) helices with a pitch of 19.0 Å (h = 6.33 Å); the second strand is translated axially relative to the first by exactly half this distance. The agreement between the observed peak positions and the cylindrically averaged Fourier transform from this left-handed model was better than the Fourier transform calculated from the right-handed double helix. Other single, double or even triple helical models having a translation period of 9.5 Å and three-fold helical symmetry were found to be sterically unacceptable. Corongiu et al. performed Monte Carlo simulations to study the hydration of this model, and concluded that no hydration site is present inside the cavity .
Foord and Atkins noted that ‘excepting the X‑ray case, evidence for the double helix is of the kind where the model fits but may not be necessary’
.
They used different experimental conditions to obtain agarose films for X‑ray diffraction. Depending on the conditions, either high crystalline forms were obtained or the fairly diffuse pattern was observed that has been interpreted by Arnott et al. as the double helix. Three highly crystalline structures were observed; two with three-fold and one with four-fold helical symmetry and with h equal to 9.73 Å, 9.38 Å, and 8.88 Å, respectively, indicating extended and single agarose chains. The occurrence of left-handed helices was calculated to be more reliable than that of right-handed models. The diffuse diffraction pattern would arise from the Fourier transform of a single-helix model (n = –3, h = 3.17 Å) ; however no stable helices with such characteristics were found.
Arndt and Stevens concluded from vacuum UV CD studies of agarose that in sols the chains are locally extended, mimicking single helices . In gels, however, the chains are less extended, mimicking wide-diameter helices, which may be intertwined in double helices. Here too, ‘the model fits but may not be necessary’ . Schafer and Stevens reexamined published optical rotation data for agarose gels by applying a recent chiroptical model of saccharide rotation . They concluded that the data are consistent with the wide-diameter helices, capable of intertwining, and not with the predominance, in the gel, of extended chains.
The glycosidic torsion angles in the models proposed by Arnott et al. and Foord and Atkins were not derived from the diffraction data, but calculated by computerized model building. Arnott et al. first calculated sterically allowed regions for the links. The final dihedrals were those that satisfied the required helical geometry and deviated minimally from the centers of these sterically allowed regions, which may deviate from energetically favorable regions. The glycosidic links in the models by Foord and Atkins were selected for similarity: the alteration in passing between one another is minimal. According to ‘a partial estimation of conformational energy’ (which is not further explained), they should be near an energetic minimum. An evaluation of the proposed models on energetical criteria is therefore of great interest .
Jimenez-Barbero et al. published a molecular modeling study of the repeating disaccharides of agarose and their relationship to agarose . In this study only isolated single helices were taken into account. According to these calculations, a left-handed three-fold helix with h = 9.5 Å was predicted to be the most stable form of the single chain, which practically coincides with two of the models given by Foord and Atkins .
The model building and simulation of the helices as proposed earlier in the literature showed that the models proposed by Foord and Atkins for the three diffraction patterns from the highly crystalline samples may very well be stable helices. As they stated themselves, a single-helical model for the diffuse diffraction pattern cannot be found. The model proposed for this pattern by Arnott et al. seems to be unstable in the 1→4 linkage.
Our search for the lowest-energy helix with helical parameters in agreement with the diffraction patterns lead to ten helical models, both left and right handed, and both single- and double-helical models for the diffuse diffraction pattern. The most favorable packing, both parallel and antiparallel of two helices of each model was then calculated. On the basis of energy and symmetry criteria, all right-handed helical models, all parallel packed left-handed helical models and for the diffuse diffraction pattern the single-helical models were rejected. Only left-handed, antiparallel packed models were left, and with these crystal structures were build, making use of the crystal systems reported by Foord and Atkins . Cell dimensions and intensities of X-ray reflections were calculated compared favorably with available experimental data.
Although the predicted crystal structure for the first diffraction pattern can not be validated with experimental information, and the structure for the last, tetragonal pattern appears to agree less with the available experimental data than the other two, it is interesting to make a comparison between the predicted structures. They have in common that all structures have antiparallel packings, and that the helices are all left handed, with minor differences in the glycosidic torsion angles and in the potential energies of the isolated helices. This facilitates transitions between the different allomorphs. They mainly differ from the models presented earlier in the literature
in the values of the y torsions in the 1→4 link. Next, all of the proposed crystal structures have enough space (30-45%) for water molecules, which is to be expected for a gel-forming polysaccharide.
The overall good agreement between the published and our calculated crystallographic data for the two trigonal crystal systems are a validation of the methods we used going from standard monosaccharide building blocks to crystal structures. Therefore, we feel it is appropriate to rely on these methods for the prediction of a crystal structure related to the first, diffuse diffraction pattern. The methods lead to a clear preference for a double helical model, packed in an anti-parallel way. In line with the construction of the two three-fold single helical crystal structures, we therefore dare to make the prediction for the crystal structure with the double helix. Thus, according to the present study, agarose appears to be capable to form both single and double helical structures.
